Laplacian inversion#
A common problem in plasma models is to solve an equation of the form
For example,
appears in reduced MHD for the vorticity inversion and \(j_{||}\).
Alternative formulations and ways to invert equation (3) can be found in section LaplaceXY and LaplaceXZ
Several implementations of the Laplacian solver are available, which are selected by changing the “type” setting.The currently available implementations are listed in table Table 12.
Name |
Description |
Requirements |
|---|---|---|
cyclic |
Serial/parallel. Gathers boundary rows onto one processor. |
|
Serial/parallel. Lots of methods, no Boussinesq |
PETSc (section PETSc) |
|
petsc3damg |
Serial/parallel. Solves full 3D operator (with y-derivatives) with algebraic multigrid. |
PETSc (section PETSc) |
multigrid |
Serial/parallel. Geometric multigrid, no Boussinesq |
|
Serial/parallel. Iterative treatment of non-Boussinesq terms |
||
Serial only. Thomas algorithm for tridiagonal system. |
Lapack (section LAPACK) |
|
Serial only. Enables 4th-order accuracy |
Lapack (section LAPACK) |
|
Parallel only (NXPE>1). Thomas algorithm. |
||
Iterative parallel tridiagonal solver. Parallel only, but automatically falls back to Thomas algorithm for NXPE=1. |
Usage of the laplacian inversion#
In BOUT++, equation (3) can be solved in two
ways. The first method Fourier transforms in the \(z\)-direction,
whilst the other solves the full two dimensional problem by matrix
inversion. The derivation of \(\nabla_\perp^2f\) for a general
coordinate system can be found in the
Field-aligned coordinates section. What is important, is to
note that if \(g_{xy}\) and \(g_{yz}\) are non-zero, BOUT++
neglects the \(y\)-parallel derivatives when using the solvers
Laplacian and LaplaceXZ.
By neglecting the \(y\)-derivatives (or if \(g_{xy}=g_{yz}=0\)), one can solve equation (3) \(y\) plane by \(y\) plane.
The first approach utilizes the fact that it is possible to Fourier transform the equation in \(z\) (using some assumptions described in section Numerical implementation), and solve a tridiagonal system for each mode. These inversion problems are band-diagonal (tri-diagonal in the case of 2nd-order differencing) and so inversions can be very efficient: \(O(n_z \log n_z)\) for the FFTs, \(O(n_x)\) for tridiagonal inversion using the Thomas algorithm, where \(n_x\) and \(n_z\) are the number of grid-points in the \(x\) and \(z\) directions respectively.
In the second approach, the full \(2\)-D system is solved. The available solvers for this approach are ‘multigrid’ using a multigrid algorithm; ‘naulin’ using an iterative scheme to correct the FFT-based approach; or ‘petsc’ using KSP linear solvers from the PETSc library (this requires PETSc to be built with BOUT++).
The Laplacian class is defined in invert_laplace.hxx and solves
problems formulated like equation (3) To use this
class, first create an instance of it:
std::unique_ptr<Laplacian> lap = Laplacian::create();
By default, this will use the options in a section called “laplace”, but
can be given a different section as an argument. By default
\(d = 1\), \(a = 0\), and \(c_1=c_2=1\). To set the values of
these coefficients, there are the setCoefA(), setCoefC1(),
setCoefC2(), setCoefC() (which sets both \(c_1\) and \(c_2\)
to its argument), and setCoefD() methods:
Field2D a = ...;
lap->setCoefA(a);
lap->setCoefC(0.5);
arguments can be Field2D, Field3D, or BoutReal values. Note that FFT
solvers will use only the DC part of Field3D arguments.
Settings for the inversion can be set in the input file under the
section laplace (default) or whichever settings section name was
specified when the Laplacian class was created. Commonly used
settings are listed in tables Table 13 to
Table 16.
In particular boundary conditions on the \(x\) boundaries can be
set using the and outer_boundary_flags variables, as detailed in
table Table 15. Note that DC (‘direct-current’)
refers to \(k = 0\) Fourier component, AC (‘alternating-current’)
refers to \(k \neq 0\) Fourier components. Non-Fourier solvers use
AC options (and ignore DC ones). Multiple boundary conditions can be
selected by adding together the required boundary condition flag
values together. For example, inner_boundary_flags = 3 will set a
Neumann boundary condition on both AC and DC components.
It is pertinent to note here that the boundary in BOUT++ is defined by
default to be located half way between the first guard point and first
point inside the domain. For example, when a Dirichlet boundary
condition is set, using inner_boundary_flags = 0 , 16, or
32, then the first guard point, \(f_{-}\) will be set to
\(f_{-} = 2v - f_+\), where \(f_+\) is the first grid point
inside the domain, and \(v\) is the value to which the boundary is
being set to.
The global_flags, inner_boundary_flags,
outer_boundary_flags and flags values can also be set from
within the physics module using setGlobalFlags,
setInnerBoundaryFlags , setOuterBoundaryFlags and
setFlags.
lap->setGlobalFlags(Global_Flags_Value);
lap->setInnerBoundaryFlags(Inner_Flags_Value);
lap->setOuterBoundaryFlags(Outer_Flags_Value);
lap->setFlags(Flags_Value);
Name |
Meaning |
Default value |
|---|---|---|
|
Which implementation to use. See table Table 12 |
|
|
Filter out modes above \((1-\) |
0 |
|
Filter modes with \(n >\) |
|
|
Include first derivative terms |
|
|
Include corrections for non-uniform meshes (dx not constant) |
Same as global |
|
Sets global inversion options See table Laplace global flags |
|
|
Sets boundary conditions on inner boundary. See table Laplace boundary flags |
|
|
Sets boundary conditions on outer boundary. See table Laplace boundary flags |
|
|
DEPRECATED. Sets global solver options and boundary conditions. See Laplace flags or invert_laplace.cxx |
|
|
Perform inversion in \(y\)-boundary guard cells |
|
Flag |
Meaning |
Code variable |
|---|---|---|
0 |
No global option set |
\(-\) |
1 |
zero DC component (Fourier solvers) |
|
2 |
set initial guess to 0 (iterative solvers) |
|
4 |
equivalent to
|
|
8 |
Use 4th order differencing (Apparently not actually implemented anywhere!!!) |
|
16 |
Set constant component (\(k_x = k_z = 0\)) to zero |
|
Flag |
Meaning |
Code variable |
|---|---|---|
0 |
Dirichlet (Set boundary to 0) |
\(-\) |
1 |
Neumann on DC component (set gradient to 0) |
|
2 |
Neumann on AC component (set gradient to 0) |
|
4 |
Zero or decaying Laplacian on AC components ( \(\frac{\partial^2}{\partial x^2}+k_z^2\) vanishes/decays) |
|
8 |
Use symmetry to enforce zero value or gradient (redundant for 2nd order now) |
|
16 |
Set boundary condition to values in boundary guard cells of second
argument, |
|
32 |
Set boundary condition to values in boundary guard cells of RHS,
|
|
64 |
Zero or decaying Laplacian on DC components (\(\frac{\partial^2}{\partial x^2}\) vanishes/decays) |
|
128 |
Assert that there is only one guard cell in the \(x\)-boundary |
|
256 |
DC value is set to parallel gradient, \(\nabla_\parallel f\) |
|
512 |
DC value is set to inverse of parallel gradient \(1/\nabla_\parallel f\) |
|
1024 |
Boundary condition for inner ‘boundary’ of cylinder |
|
Flag |
Meaning |
|---|---|
1 |
Zero-gradient DC on inner (X) boundary. Default is zero-value |
2 |
Zero-gradient AC on inner boundary |
4 |
Zero-gradient DC on outer boundary |
8 |
Zero-gradient AC on outer boundary |
16 |
Zero DC component everywhere |
32 |
Not used currently |
64 |
Set width of boundary to 1 (default is |
128 |
Use 4\(^{th}\)-order band solver (default is 2\(^{nd}\) order tridiagonal) |
256 |
Attempt to set zero laplacian AC component on inner boundary by combining 2nd and 4th-order differencing at the boundary. Ignored if tridiagonal solver used. |
512 |
Zero laplacian AC on outer boundary |
1024 |
Symmetric boundary condition on inner boundary |
2048 |
Symmetric outer boundary condition |
To perform the inversion, there’s the solve method
x = lap->solve(b);
There are also functions compatible with older versions of the BOUT++ code, but these are deprecated:
Field2D a, c, d;
invert_laplace(b, x, flags, &a, &c, &d);
and
x = invert_laplace(b, flags, &a, &c, &d);
The input b and output x are 3D fields, and the coefficients
a, c, and d are pointers to 2D fields. To omit any of the
three coefficients, set them to NULL.
Numerical implementation#
We will here go through the implementation of the laplacian inversion algorithm, as it is performed in BOUT++. We would like to solve the following equation for \(f\)
BOUT++ neglects the \(y\)-parallel derivatives if \(g_{xy}\)
and \(g_{yz}\) are non-zero when using the solvers Laplacian and
LaplaceXZ. For these two solvers, equation (4) becomes
(see Field-aligned coordinates for derivation)
Using tridiagonal solvers#
Since there are no parallel \(y\)-derivatives if \(g_{xy}=g_{yz}=0\) (or if they are neglected), equation (4) will only contain derivatives of \(x\) and \(z\) for the dependent variable. The hope is that the modes in the periodic \(z\) direction will decouple, so that we in the end only have to invert for the \(x\) coordinate.
If the modes decouples when Fourier transforming equation (5), we can use a tridiagonal solver to solve the equation for each Fourier mode.
Using the discrete Fourier transform
we see that the modes will not decouple if a term consist of a product of two terms which depends on \(z\), as this would give terms like
Thus, in order to use a tridiagonal solver, \(a\), \(c_1\), \(c_2\) and \(d\) cannot be functions of \(z\). Because of this, the \({{\boldsymbol{e}}}^z \partial_z c_2\) term in equation (5) is zero. Thus the tridiagonal solvers solve equations of the form
after using the discrete Fourier transform (see section Derivatives of the Fourier transform), we get
which gives
As nothing in equation (6) couples points in \(y\) together (since we neglected the \(y\)-derivatives if \(g_{xy}\) and \(g_{yz}\) were non-zero) we can solve \(y\)-plane by \(y\)-plane. Also, as the modes are decoupled, we may solve equation (6) \(k\) mode by \(k\) mode in addition to \(y\)-plane by \(y\)-plane.
The second order centred approximation of the first and second derivatives in \(x\) reads
This gives
collecting point by point
We now introduce
which inserted in equation (7) gives
This can be formulated as the matrix equation
where the matrix \(A\) is tridiagonal. The boundary conditions are set by setting the first and last rows in \(A\) and \(B_z\).
The tridiagonal solvers previously required \(c_1 = c_2\) in equation (4), but from version 4.3 allow \(c_1 \neq c_2\).
Using PETSc solvers#
When using PETSc, all terms of equation (5) are used when inverting to find \(f\). Note that when using PETSc, we do not Fourier decompose in the \(z\)-direction, so it may take substantially longer time to find the solution. As with the tridiagonal solver, the fields are sliced in the \(y\)-direction, and a solution is found for one \(y\) plane at the time.
Before solving, equation (5) is rewritten to the form \(A{{\boldsymbol{x}}} ={{\boldsymbol{b}}}\) (however, the full \(A\) is not expanded in memory). To do this, a row \(i\) in the matrix \(A\) is indexed from bottom left of the two dimensional field \(= (0,0) = 0\) to top right \(= (\texttt{meshx}-1, \texttt{meshz}-1) = \texttt{meshx}\cdot\texttt{meshz}-1\) of the two dimensional field. This is done in such a way so that a row \(i\) in \(A\) increments by \(1\) for an increase of \(1\) in the \(z-\)direction, and by \(\texttt{meshz}\) for an increase of \(1\) in the \(x-\)direction, where the variables \(\texttt{meshx}\) and \(\texttt{meshz}\) represents the highest value of the field in the given direction.
Similarly to equation (7), the discretised version of equation (5) can be written. Doing the same for the full two dimensional case yields:
Second order approximation
Fourth order approximation
To determine the coefficient for each node point, it is convenient to introduce some quantities
In addition, we have:
Second order approximation (5-point stencil)
Fourth order approximation (9-point stencil)
This gives
The coefficients \(c_{i+m,j+n}\) are finally being set according
to the appropriate order of discretisation. The coefficients can be
found in the file petsc_laplace.cxx.
Example: The 5-point stencil#
Let us now consider the 5-point stencil for a mesh with \(3\) inner points in the \(x\)-direction, and \(3\) inner points in the \(z\)-direction. The \(z\) direction will be periodic, and the \(x\) direction will have the boundaries half between the grid-point and the first ghost point (see Fig. 11).
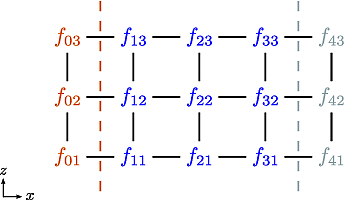
Fig. 11 The mesh: The inner boundary points in \(x\) are coloured in orange, whilst the outer boundary points in \(z\) are coloured gray. Inner points are coloured blue.#
Applying the \(5\)-point stencil to point \(f_{22}\) this mesh will result in Fig. 12.
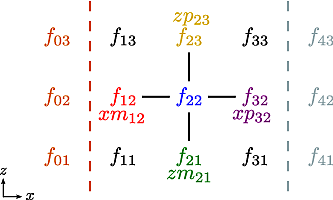
Fig. 12 The mesh with a stencil in point \(f_{22}\): The point under
consideration is coloured blue. The point located \(+1\) in
\(z\) direction (zp) is coloured yellow and the point
located \(-1\) in \(z\) direction (zm) is coloured
green. The point located \(+1\) in \(x\) direction (xp)
is coloured purple and the point located \(-1\) in \(x\)
direction (xm) is coloured red.#
We want to solve a problem on the form \(A{{\mathbf{x}}}={{\mathbf{b}}}\). We will order \({{\mathbf{x}}}\) in a row-major order (so that \(z\) is varying faster than \(x\)). Further, we put the inner \(x\) boundary points first in \({{\mathbf{x}}}\), and the outer \(x\) boundary points last in \({{\mathbf{x}}}\). The matrix problem for our mesh can then be written like in Fig. 13.

Fig. 13 Matrix problem for our \(3\times3\) mesh: The colors follow
that of figure Fig. 11 and
Fig. 12. The first index of the
elements refers to the \(x\)-position in figure
Fig. 11, and the last index of the elements
refers to the \(z\)-position in figure
Fig. 11. ig refers to “inner ghost point”,
og refers to “outer ghost point”, and c refers to the point
of consideration. Notice the “wrap-around” in \(z\)-direction
when the point of consideration neighbours the first/last
\(z\)-index.#
As we are using a row-major implementation, the global indices of the matrix will be as in Fig. 14

Implementation internals#
The Laplacian inversion code solves the equation:
where \(x\) and \(b\) are 3D variables, whilst \(a\), \(c_1\), \(c_2\) and \(d\) are 2D variables for the FFT solvers, or 3D variables otherwise. Several different algorithms are implemented for Laplacian inversion, and they differ between serial and parallel versions. Serial inversion can currently either be done using a tridiagonal solver (Thomas algorithm), or a band-solver (allowing \(4^{th}\)-order differencing).
To support multiple implementations, a base class Laplacian is
defined in include/invert_laplace.hxx. This defines a set of
functions which all implementations must provide:
class Laplacian {
public:
virtual void setCoefA(const Field2D &val) = 0;
virtual void setCoefC(const Field2D &val) = 0;
virtual void setCoefD(const Field2D &val) = 0;
virtual const FieldPerp solve(const FieldPerp &b) = 0;
}
At minimum, all implementations must provide a way to set coefficients, and a solve function which operates on a single FieldPerp (X-Y) object at once. Several other functions are also virtual, so default code exists but can be overridden by an implementation.
For convenience, the Laplacian base class also defines a function to
calculate coefficients in a Tridiagonal matrix:
void tridagCoefs(int jx, int jy, int jz, dcomplex &a, dcomplex &b,
dcomplex &c, const Field2D *c1coef = nullptr,
const Field2D *c2coef = nullptr,
const Field2D *d=nullptr);
For the user of the class, some static functions are defined:
static std::unique_ptr<Laplacian> create(Options *opt = nullptr);
static Laplacian* defaultInstance();
The create function allows new Laplacian implementations to be created,
based on options. To use the options in the [laplace] section, just
use the default:
std::unique_ptr<Laplacian> lap = Laplacian::create();
The code for the Laplacian base class is in
src/invert/laplace/invert_laplace.cxx. The actual creation of new
Laplacian implementations is done in the LaplaceFactory class,
defined in src/invert/laplace/laplacefactory.cxx. This file
includes all the headers for the implementations, and chooses which
one to create based on the type setting in the input options. This
factory therefore provides a single point of access to the underlying
Laplacian inversion implementations.
Each of the implementations is in a subdirectory of
src/invert/laplace/impls and is discussed below.
Serial tridiagonal solver#
This is the simplest implementation, and is in
src/invert/laplace/impls/serial_tri/
Serial band solver#
This is band-solver which performs a \(4^{th}\)-order inversion.
Currently this is only available when NXPE=1; when more than one
processor is used in \(x\), the Laplacian algorithm currently
reverts to \(3^{rd}\)-order.
SPT parallel tridiagonal#
This is a reference code which performs the same operations as the
serial code. To invert a single XZ slice (FieldPerp object), data
must pass from the innermost processor (mesh->PE_XIND = 0) to the
outermost mesh->PE_XIND = mesh->NXPE-1 and back again.
Some parallelism is achieved by running several inversions
simultaneously, so while processor 1 is inverting Y=0, processor 0 is
starting on Y=1. This works ok as long as the number of slices to be
inverted is greater than the number of X processors
(MYSUB > mesh->NXPE). If MYSUB < mesh->NXPE then not all
processors can be busy at once, and so efficiency will fall sharply.
Fig. 15 shows the useage of 4 processors inverting a
set of 3 poloidal slices (i.e. MYSUB=3)
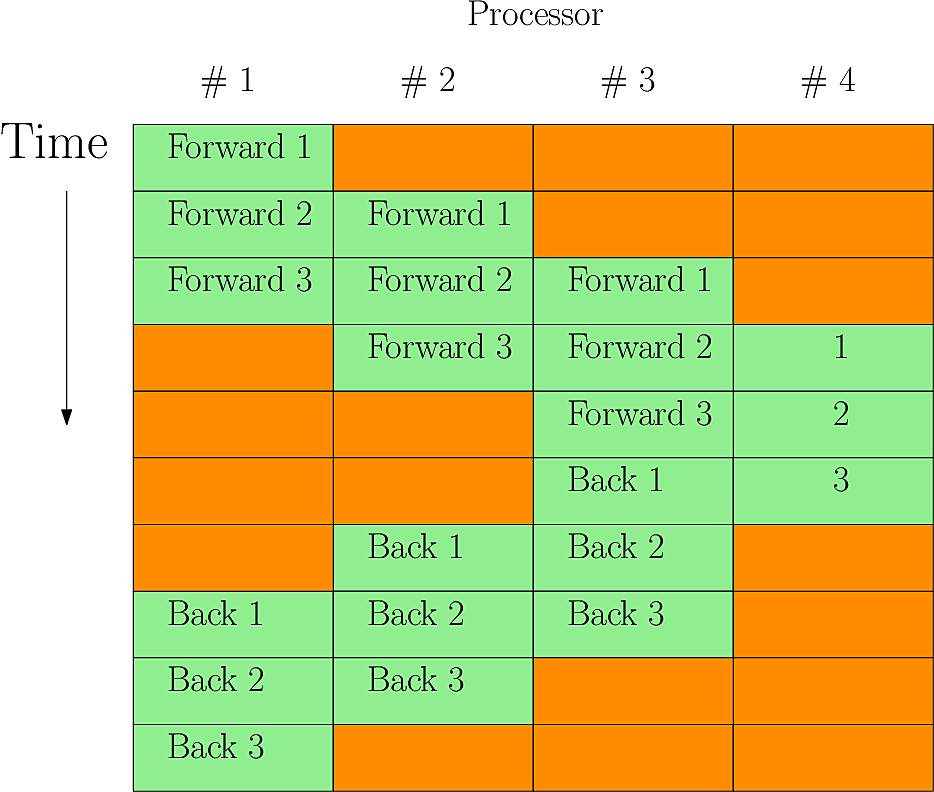
Fig. 15 Parallel Laplacian inversion with MYSUB=3 on 4 processors. Red periods are where a processor is idle - in this case about 40% of the time#
Cyclic algorithm#
This is now the default solver in both serial and parallel. It is an FFT-based solver using a cyclic reduction algorithm.
Multigrid solver#
A solver using a geometric multigrid algorithm was introduced by projects in 2015 and 2016 of CCFE and the EUROfusion HLST.
Naulin solver#
This scheme was introduced for BOUT++ by Michael Løiten in the CELMA code and the iterative algorithm is detailed in his thesis [Løiten2017].
The iteration can be under-relaxed (see naulin_laplace.cxx for more details of the
implementation). A factor \(0< \text{underrelax\_factor}<=1\) is used, with a value of 1 corresponding
to no under-relaxation. If the iteration starts to diverge (the error increases on any
step) the underrelax_factor is reduced by a factor of 0.9, and the iteration is restarted
from the initial guess. The initial value of underrelax_factor, which underrelax_factor is
set to at the beginning of each call to solve can be set by the option
initial_underrelax_factor (default is 1.0) in the appropriate section of the input
file ([laplace] by default). Reducing the value of initial_underrelax_factor may
speed up convergence in some cases. Some statistics from the solver are written to the
output files to help in choosing this value. With <i> being the number of the
LaplaceNaulin solver, counting in the order they are created in the physics model:
naulinsolver<i>_mean_underrelax_countsgives the mean number of timesunderrelax_factorhad to be reduced to get the iteration to converge. If this is much above 0, it is probably worth reducinginitial_underrelax_factor.naulinsolver<i>_mean_itsis the mean number of iterations taken to converge. Try to minimise when adjustinginitial_underrelax_factor.
Michael Løiten, “Global numerical modeling of magnetized plasma in a linear device”, 2017, https://celma-project.github.io/.
Iterative Parallel Tridiagonal solver#
This solver uses a hybrid of multigrid and the Thomas algorithm to invert
tridiagonal matrices in parallel.
The complexity of the algorithm is O(nx) work and O(log(NXPE))
communications.
The Laplacian is second-order, so to invert it we need two boundary conditions, one at each end of the domain. If we only have one processor, that processor knows both boundary conditions, and we can invert the Laplacian locally using the Thomas algorithm. When the domain is subdivided between two or more processors, we can no longer use the Thomas algorithm, as processors do not know the solution at the subdomain boundaries.
In this hybrid approach, we reduce the original system of equations to a smaller system for solution at the boundaries of each processor’s subdomain. We solve this system in parallel using multigrid. Once the boundary values are known, each processor can find the solution on its subdomain using the Thomas algorithm.
Parameters.
type = iptselects this solver.rtolandatolare the relative and absolute error tolerances to determine when the residual has converged. The goal of setting these is to minimize the runtime by minimizing the number of iterations required to meet the tolerance. Intuitively we would expect tightening tolerances is bad, as doing so requires more iterations. This is true, but also loosening the tolerances too far can lead to very slowly convergence. Generally, as we scan from large to small tolerances, we start with very slow calculations, then meet some threshold in tolerance where the runtime drops sharply, then see runtime slowly increase again as tolerances tighten further. The run time for all tolerances below this threshold are similar though, and generally it is best to err on the side of tighter tolerances.maxitsis the maximum number of iterations allowed before the job fails.max_cycleis the number of pre and post smoothing operations applied on each multigrid level. The optimal value appears to bemax_cycle = 1.max_levelsets the number of multigrid levels. The optimal value is usually the largest possible valuemax_level = log2(NXPE) - 2(see “constraints” below), but sometimes one or two levels less than this can be faster.predict_exit. Multigrid convergence rates are very robust. Whenpredict_exit = true, we calculate the convergence rate from early iterations and predict the iteration at which the algorithm will have converged. This allows us to skip convergence checks at most iterations (these checks are expensive as they require global communication). Whether this is advantageous is problem-dependent: it is probably useful at lowZresolution but not at higherZresolution. This is because the algorithm skips work associated withkzmodes which have converged; but if we do not check convergence, we do not know which modes we can skip. Therefore at higherZresolution we find that reduced communication costs are offset by increased work.predict_exitdefaults tofalse.
Constraints. This method requires that:
NXPEis a power of 2.NXPE > 2^(max_levels+1)
LaplaceXY#
Perpendicular Laplacian solver in X-Y.
In 2D (X-Y), the \(g_{xy}\) component can be dropped since this depends on integrated shear \(I\) which will cancel with the \(g_{xz}\) component. The \(z\) derivative is zero and so this simplifies to
The divergence operator in conservative form is
and so the perpendicular Laplacian in X-Y is
In field-aligned coordinates, the metrics in the \(y\) derivative term become:
In the LaplaceXY operator this is implemented in terms of fluxes at cell faces.
Notes:
The
ShiftedMetricorFCITransformParallelTransform must be used (i.e.mesh:paralleltransform:type = shiftedormesh:paralleltransform:type = fci) for this to work, since it assumes that \(g^{xz} = 0\)Setting the option
pctype = hypreseems to work well, if PETSc has been compiled with the algebraic multigrid library hypre; this can be included by passing the option--download-hypreto PETSc’sconfigurescript.LaplaceXY(with the default finite-volume discretisation) has a slightly different convention for passing non-zero boundary values than theLaplaciansolvers.LaplaceXYuses the average of the last grid cell and first boundary cell of the initial guess (second argument tosolve()) as the value to impose for the boundary condition.
An alternative discretization is available if the option finite_volume =
false is set. Then a finite-difference discretization very close to the one used when
calling A*Laplace_perp(f) + Grad_perp(A)*Grad_perp(f) + B*f is used. This also
supports non-orthogonal grids with \(g^{xy} \neq 0\). The difference is that when
\(g^{xy} \neq 0\), Laplace_perp calls D2DXDY(f) which applies a boundary
condition to dfdy = DDY(f) before calculating DDX(dfdy) with a slightly different
result than the way boundary conditions are applied in LaplaceXY.
The finite difference implementation of
LaplaceXYpasses non-zero values for the boundary conditions in the same way as theLaplaciansolvers. The value in the first boundary cell of the initial guess (second argument tosolve()) is used as the boundary value. (Note that this value is imposed as a boundary condition on the returned solution at a location half way between the last grid cell and first boundary cell.)
LaplaceXZ#
This is a Laplacian inversion code in X-Z, similar to the
Laplacian solver described in Laplacian inversion. The
difference is in the form of the Laplacian equation solved, and the
approach used to derive the finite difference formulae. The equation
solved is:
where \(A\) and \(B\) are coefficients, \(b\) is the known
RHS vector (e.g. vorticity), and \(f\) is the unknown quantity to
be calculated (e.g. potential), and \(\nabla_\perp f\) is the same
as equation ((8)), but with negligible
\(y\)-parallel derivatives if \(g_{xy}\), \(g_{yz}\) and
\(g_{xz}\) is non-vanishing. The Laplacian is written in
conservative form like the LaplaceXY solver, and
discretised in terms of fluxes through cell faces.
The header file is include/bout/invert/laplacexz.hxx. The solver is
constructed by using the LaplaceXZ::create() function:
std::unique_ptr<LaplaceXZ> lap = LaplaceXZ::create(mesh);
Note that a pointer to a Mesh object must be given, which
for now is the global variable mesh. By default the
options section laplacexz is used, so to set the type of solver
created, set in the options
[laplacexz]
type = petsc # Set LaplaceXZ type
or on the command-line laplacexz:type=petsc .
The coefficients must be set using setCoefs . All coefficients must
be set at the same time:
lap->setCoefs(1.0, 0.0);
Constants, Field2D or Field3D values can be passed. If the
implementation doesn’t support Field3D values then the average over
\(z\) will be used as a Field2D value.
To perform the inversion, call the solve function:
Field3D vort = ...;
Field3D phi = lap->solve(vort, 0.0);
The second input to solve is an initial guess for the solution,
which can be used by iterative schemes e.g. using PETSc.
Implementations#
The currently available implementations are:
cyclic: This implementation assumes coefficients are constant in \(Z\), and uses FFTs in \(z\) and a complex tridiagonal solver in \(x\) for each \(z\) mode (theCyclicReductionsolver). Code insrc/invert/laplacexz/impls/cyclic/.petsc: This uses the PETSc KSP interface to solve a matrix with coefficients varying in both \(x\) and \(z\). To improve efficiency of direct solves, a different matrix is used for preconditioning. When the coefficients are updated the preconditioner matrix is not usually updated. This means that LU factorisations of the preconditioner can be re-used. Since this factorisation is a large part of the cost of direct solves, this should greatly reduce the run-time.
Test case#
The code in examples/test-laplacexz is a simple test case for
LaplaceXZ . First it creates a LaplaceXZ
object:
std::unique_ptr<LaplaceXZ> inv = LaplaceXZ::create(mesh);
For this test the petsc implementation is the default:
[laplacexz]
type = petsc
ksptype = gmres # Iterative method
pctype = lu # Preconditioner
By default the LU preconditioner is used. PETSc’s built-in factorisation only works in serial, so for parallel solves a different package is needed. This is set using:
factor_package = superlu_dist
This setting can be “petsc” for the built-in (serial) code, or one of “superlu”, “superlu_dist”, “mumps”, or “cusparse”.
Then we set the coefficients:
inv->setCoefs(Field3D(1.0),Field3D(0.0));
Note that the scalars need to be cast to fields (Field2D or Field3D)
otherwise the call is ambiguous. Using the PETSc command-line flag
-mat_view ::ascii_info information on the assembled matrix is
printed:
$ mpirun -np 2 ./test-laplacexz -mat_view ::ascii_info
...
Matrix Object: 2 MPI processes
type: mpiaij
rows=1088, cols=1088
total: nonzeros=5248, allocated nonzeros=5248
total number of mallocs used during MatSetValues calls =0
not using I-node (on process 0) routines
...
which confirms that the matrix element pre-allocation is setting the correct number of non-zero elements, since no additional memory allocation was needed.
A field to invert is created using FieldFactory:
Field3D rhs = FieldFactory::get()->create3D("rhs",
Options::getRoot(),
mesh);
which is currently set to a simple function in the options:
rhs = sin(x - z)
and then the system is solved:
Field3D x = inv->solve(rhs, 0.0);
Using the PETSc command-line flags -ksp_monitor to monitor the
iterative solve, and -mat_superlu_dist_statprint to monitor
SuperLU_dist we get:
Nonzeros in L 19984
Nonzeros in U 19984
nonzeros in L+U 38880
nonzeros in LSUB 11900
NUMfact space (MB) sum(procs): L\U 0.45 all 0.61
Total highmark (MB): All 0.62 Avg 0.31 Max 0.36
Mat conversion(PETSc->SuperLU_DIST) time (max/min/avg):
4.69685e-05 / 4.69685e-05 / 4.69685e-05
EQUIL time 0.00
ROWPERM time 0.00
COLPERM time 0.00
SYMBFACT time 0.00
DISTRIBUTE time 0.00
FACTOR time 0.00
Factor flops 1.073774e+06 Mflops 222.08
SOLVE time 0.00
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 28.67
0 KSP Residual norm 5.169560044060e+02
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 60.50
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 49.86
1 KSP Residual norm 1.359142853145e-12
So after the initial setup and factorisation, the system is solved in one iteration using the LU direct solve.
As a test of re-using the preconditioner, the coefficients are then modified:
inv->setCoefs(Field3D(2.0),Field3D(0.1));
and solved again:
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 84.15
0 KSP Residual norm 5.169560044060e+02
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 90.42
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 98.51
1 KSP Residual norm 2.813291076609e+02
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 94.88
2 KSP Residual norm 1.688683980433e+02
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 87.27
3 KSP Residual norm 7.436784980024e+01
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 88.77
4 KSP Residual norm 1.835640800835e+01
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 89.55
5 KSP Residual norm 2.431147365563e+00
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 88.00
6 KSP Residual norm 5.386963293959e-01
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 93.50
7 KSP Residual norm 2.093714782067e-01
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 91.91
8 KSP Residual norm 1.306701698197e-02
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 89.44
9 KSP Residual norm 5.838501185134e-04
SOLVE time 0.00
Solve flops 8.245800e+04 Mflops 81.47
Note that this time there is no factorisation step, but the direct solve is still very effective.
Blob2d comparison#
The example examples/blob2d-laplacexz is the same as
examples/blob2d but with LaplaceXZ rather than
Laplacian.
Tests on one processor: Using Boussinesq approximation, so that the matrix elements are not changed, the cyclic solver produces output:
1.000e+02 125 8.28e-01 71.8 8.2 0.4 0.6 18.9
2.000e+02 44 3.00e-01 69.4 8.1 0.4 2.1 20.0
whilst the PETSc solver with LU preconditioner outputs:
1.000e+02 146 1.15e+00 61.9 20.5 0.5 0.9 16.2
2.000e+02 42 3.30e-01 58.2 20.2 0.4 3.7 17.5
so the PETSc direct solver seems to take only slightly longer than the cyclic solver. For comparison, GMRES with Jacobi preconditioning gives:
1.000e+02 130 2.66e+00 24.1 68.3 0.2 0.8 6.6
2.000e+02 78 1.16e+00 33.8 54.9 0.3 1.1 9.9
and with SOR preconditioner:
1.000e+02 124 1.54e+00 38.6 50.2 0.3 0.4 10.5
2.000e+02 45 4.51e-01 46.8 37.8 0.3 1.7 13.4
When the Boussinesq approximation is not used, the PETSc solver with LU preconditioning, re-setting the preconditioner every 100 solves gives:
1.000e+02 142 3.06e+00 23.0 70.7 0.2 0.2 6.0
2.000e+02 41 9.47e-01 21.0 72.1 0.3 0.6 6.1
i.e. around three times slower than the Boussinesq case. When using jacobi preconditioner:
1.000e+02 128 2.59e+00 22.9 70.8 0.2 0.2 5.9
2.000e+02 68 1.18e+00 26.5 64.6 0.2 0.6 8.1
For comparison, the Laplacian solver using the
tridiagonal solver as preconditioner gives:
1.000e+02 222 5.70e+00 17.4 77.9 0.1 0.1 4.5
2.000e+02 172 3.84e+00 20.2 74.2 0.2 0.2 5.2
or with Jacobi preconditioner:
1.000e+02 107 3.13e+00 15.8 79.5 0.1 0.2 4.3
2.000e+02 110 2.14e+00 23.5 69.2 0.2 0.3 6.7
The LaplaceXZ solver does not appear to be dramatically faster in
serial than the Laplacian solver when the matrix coefficients are
modified every solve. When matrix elements are not modified then the
solve time is competitive with the tridiagonal solver.
As a test, timing only the setCoefs call for the non-Boussinesq case
gives:
1.000e+02 142 1.86e+00 83.3 9.5 0.2 0.3 6.7
2.000e+02 41 5.04e-01 83.1 8.0 0.3 1.2 7.3
so around 9% of the run-time is in setting the coefficients, and the remaining \(\sim 60\)% in the solve itself.